A Fast and Quantitative Method for Post-translational Modification and Variant Enabled Mapping of Peptides to Genomes
Графічне зображення, підкресливши, в якій застосовується стадії робочого процесу регулярних proteomic ПОГО18, а також нижче за течією параметри візуалізації, показаний на малюнку 5. Рушниця протеоміки (тобто, протеолітичних перетравлення білків, слідують рідинної хроматографії в поєднанні з тандем мас-спектрометрії) є один крок коляска proteogenomic зіставлення. Внаслідок масового спектри тандем зазвичай порівнюються з Теоретичні спектри, похідні від баз даних послідовності білка. Протеогеномікі дослідження ввести переклад послідовності Роман стенограми з кодування потенціал і не є синонімами одиничних нуклеотидних варіанти (SNVs) в базу даних, що робить його важко співвіднести ці назад в геном ссилка8. Графічний інтерфейс користувача пого (PoGoGUI) підтримує формати файлів для стандартизованої звітності пептид ідентифікації від мас-спектрометрії експериментів і перетворює їх в формат спрощеної 4-колонки pogo. PoGoGUI служить оболонкою для програми командного рядка пого і таким чином дозволяє відображення пептидів на геном координати, використовуючи посилання анотації білка кодування генів, зазвичай передбачено в ГЦФ і послідовності перекладений текст у форматі FASTA. Різних форматах генеруються PoGo можливість візуалізації різних аспектів пептиди, виявлених за допомогою мас-спектрометрії, в тому числі стовп-поступальні зміни і пептиду рівня кількісна оцінка. Вихідні файли в ліжку можна далі перетворюється і об'єднані в онлайн доступні каталоги, називається трек концентратори. Один вихідні файли, а також трек концентратори, потім можуть бути візуалізовані в браузерах, таких як браузер геномі UCSC25, Ensembl генома браузер20, IGV24і Biodalliance28 (див. Нижній Малюнок 5).
Ми застосували PoGo до реаналіза проект протеома людини карти фільтрують на велике значення, як описано в Райт та ін. 7 і порівняв його з двох інших інструментів для proteogenomic карт, а саме группе14 і PGx10. Набір даних складається з 233,055 унікальний пептиди через 59 дорослих і плода тканини, що призводить до в цілому більше 3 мільйонів послідовностями. Пого перевищили ці інструменти в середовищі виконання (6,9 x і 96,4 x швидше, відповідно) і використання пам'яті (20% і 60% менше пам'яті, відповідно) як показано на малюнку 6 18. На малюнку 7 наведено приклад успішно співставленим пептиду .
У той час як пого значно перевершує інші інструменти в швидкості і пам'яті, він також здатний стовп-поступальні зміни зіставлення і кількісної інформації, пов'язаної з пептидами на геном. Мал. 8А схематично зображує візуалізації ліжка формату в браузері генома для пептидів, зіставлення один Екзон і через зрощування розв'язок. Пого використовує варіант розмальовки для надання легко візуальна щодо унікальність пептид картування в геномі. Зіставлення в червоному вказують неповторність єдиного стенограма, а чорні моменти зіставлення одного гена. Однак пептид розподіляється між різними стенограм. Сірий зіставлення показують пептид спільно між кількома генами. Такі, наприклад, менш надійною кількісної оцінки гена або ненадійним для виклику вираження гена. PTM ліжко варіант PoGo переопределяет колірний код для розміщення різних типів стовп-поступальні зміни, як показано на Малюнок 8B. Крім того PTMs позначаються блоків товщиною (див. Малюнок 8B). Один PTM типу виділений блок товщиною в позиції модифікованих амінокислотних залишків, в той час як кілька PTMs того ж типу займаних товщиною блоку від перших модифікованих аміно кислоти до останнього.
Ми застосували пого і згодом TrackHubGenerator в набір 50 колоректальний рак клітинних ліній, включаючи весь протеома і phosphoproteome29. Хоча трек хаб, завантаженої в браузері геномі UCSC показує пептиди, зіставляються з геном і підкреслює унікальність зіставлення і фосфорилювання сайти (див. Рис. 9), додаткові дані надаються додаткові папки. GCT файли потім включите візуалізації пептиду і phosphopeptide кількісний в геномної контексті. Однак файли GCT не забезпечують простий візуалізації пептидів, що охоплюють через з'єднувач розв'язок (див. Рис. 10 Топ). Пептиди через з'єднувач розв'язок розділені на їх відповідних частин зіставлення екзонів. Хоча це можна визначити з'єднувач пептиди через ті ж кількісні значення екзона зіставлень, завантаження зіставлень на основі послідовності файлів наприклад, ліжко або GTF, підключіть екзонів, тонкий Интрон, що охоплюють лінію підтримки тлумачення (див. Малюнок 10 внизу).
Щоб підкреслити корисність варіант включено зіставлення, ми застосували пого в двох конфігураціях для dataset протеома людини яєчок, Пошук проти neXtProt полювати за відсутніх білків, з використанням декількох ферментних стратегіі22. NeXtProt включає в себе крім посилання білкових послідовностей понад 5 мільйонів однієї амінокислоти варіанти30. Зіставлення пептиди, ототожнюється з однією амінокислоти варіант не підтримується іншими засобами відображення. В цілому 177,012 унікальний пептиди були визначені. З них пептиди 99,8% (176,694) були вперше успішно зіставлені не дозволяючи невідповідності. Видалення зі списку виявлених пептид привели до 0,2% (318) пептидів, які згодом були зіставлених дозволяючи однієї амінокислоти заміни. Це призвело до 3.446 зіставлення 162 пептидів, що не були зіставлені з геном посилання з інших доступних інструментів. У той час як середня кількість зіставлень, включаючи невідповідність є високим, 62 пептиди були зіставлені тільки один локус, яке вказує істинний варіант послідовності. Приклад пептиду, зіставлених з однієї амінокислоти заміни виділяється з його послідовності і перекладеного геномні послідовності на малюнку 11.
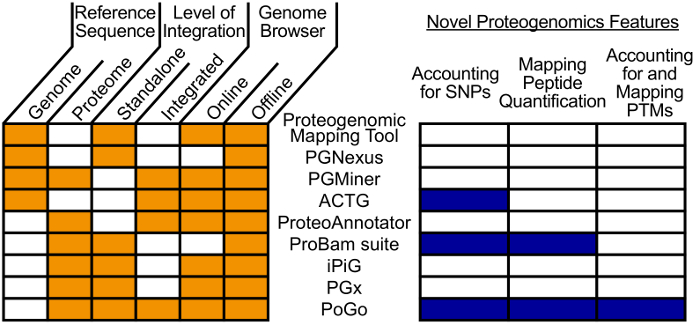
Малюнок 1. Візуальне порівняння інструментів різних пептид геном картографування. Що стосується різних аспектів показано порівняння. Ці аспекти включають в себе посилання на зіставлення, рівень інтеграції в рамки і підтримку онлайн і оффлайн браузери. Крім того нові аспекти Протеогеномікі і їх функція підтримки виділяється окремо. Пого не вистачає тільки можливість безпосередньо зіставляються генома, в порівнянні з іншими інструментами. Однак він підтримує всі нових функцій, які не підтримують більшість інших інструментів. Будь ласка, натисніть тут, щоб подивитися велику версію цієї фігури.

Малюнок 2. Приклад введення файлу для зіставлення пептиди. Пого приймає вхідні дані в форматі табуляцією з 4 колонками. Заголовки стовпців у першому рядку є «Експеримент», «Пептид», «PSMs» і «Квант», який вказує в наступних рядках експеримент або зразок ідентифікатора, послідовність пептид, кількість матчів пептид спектра і кількісне значення для пептиду, відповідно. Підтримувані розширення імен файлів * .txt, * .tsv і * .pogo. Будь ласка, натисніть тут, щоб подивитися велику версію цієї фігури.
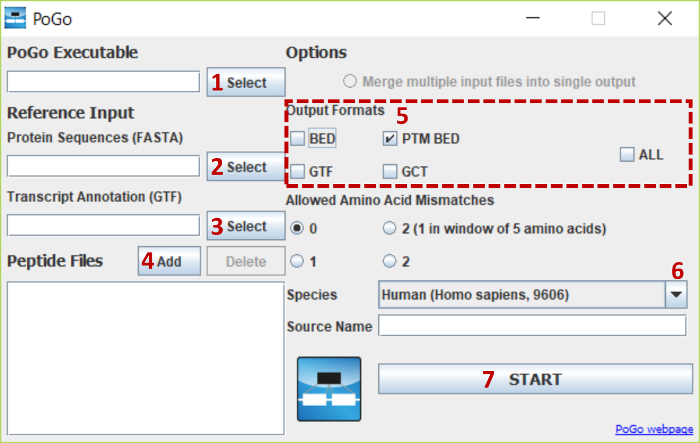
Малюнок 3. PoGoGUI інтерфейс з виділених кроків для вибору файлів і настройки параметрів. На малюнку показані кроки для вибору і завантаження всіх необхідних файлів і вибір варіантів для зіставлення пептиди з стовп-поступальні зміни на геном людини посилання. Будь ласка, натисніть тут, щоб подивитися велику версію цієї фігури.
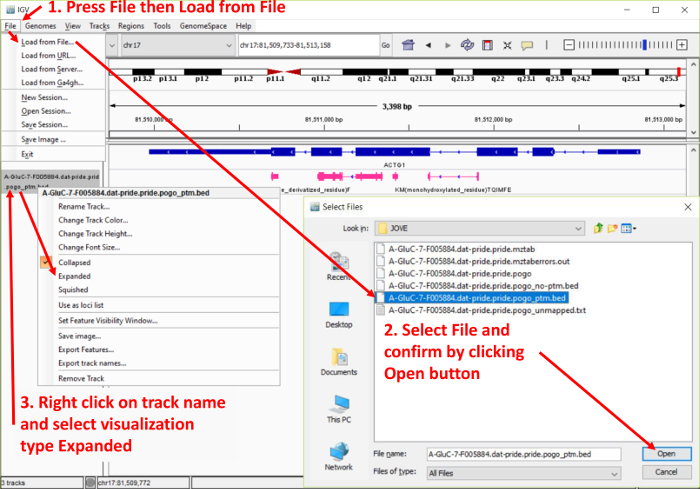
Малюнок 4. Скріншот інтегративної геноміки перегляду (IGV) даних додано процедури. Фігура висвітлює кроки, необхідні для завантаження пого вихідних файлів в браузері IGV. Крім того він показує параметр розширення трек зіставлених пептиди, щоб виділити картування і послідовності. Будь ласка, натисніть тут, щоб подивитися велику версію цієї фігури.
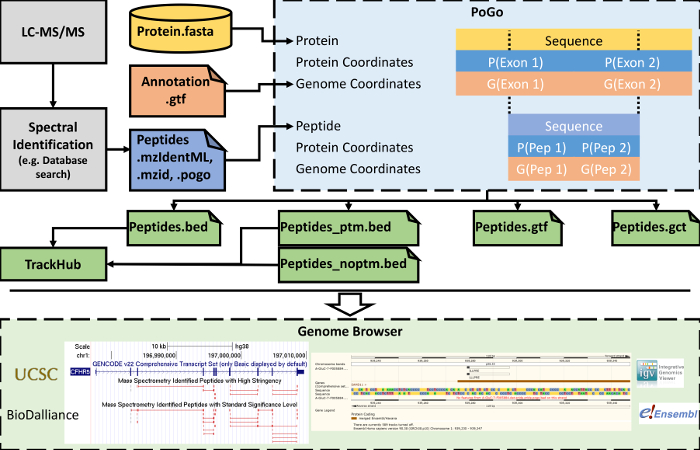
Малюнок 5. Спрощений процес візуалізації в геном браузерів в декількох кроках від LC-MS / MS. Пого зіставлення слід ідентифікація пептидів з маси спектри тандем. Для досягнення картування геному, пого використовує посилання анотації, як Анотація геномів (ГЦФ) і Стенограма переклад послідовності (FASTA). Різні формати створюються виведення, який може бути завантажений окремо в геном браузерів. Крім того файли в форматі ліжка можуть бути об'єднані в трек центрів підтримки візуалізації великих наборів даних. Будь ласка, натисніть тут, щоб подивитися велику версію цієї фігури.
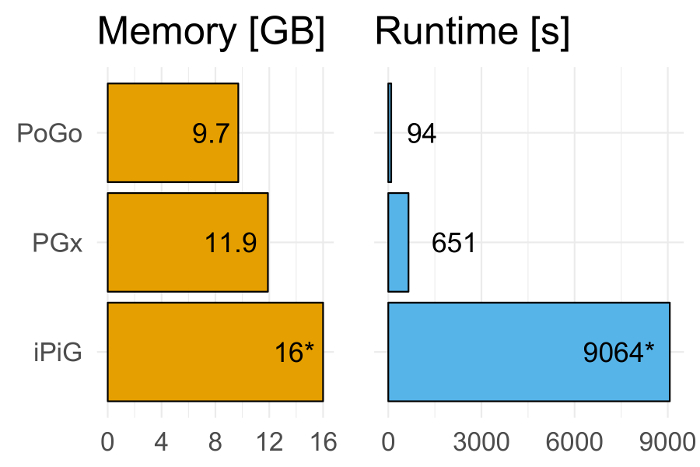
Малюнок 6. Бенчмаркінг пого проти PGx і групі. Пого перевершує інші інструменти на бенчмаркінг. Картування 233,055 унікальний пептиди через 59 дорослих і плода тканини, що призводить до більш 3 мільйонів послідовностями, пого була 6,9 x і 96,4 x швидше, ніж PGx і групі, відповідно. Крім того пого потрібно 20% і 60% менше пам'яті порівняно з PGx і групі, відповідно. У той час як пого і PGx завершився успішно, групі привели до помилка пам'яті в 16 ГБ. Будь ласка, натисніть тут, щоб подивитися велику версію цієї фігури.

Малюнок 7. Геном UCSC браузера приклад уявлення зіставлених пептидів. На малюнку показана пептиди, зіставляються mTOR ген. У той час як комбіновані трек показує пептиди, що охоплюють через з'єднувач розв'язок і зіставлення тільки один Екзон з пов'язані послідовності, тканини конкретні треки тільки виділити зіставлення в стислий формат. Будь ласка, натисніть тут, щоб подивитися велику версію цієї фігури.

Малюнок 8. Схема зіставлення візуалізації і колірне кодування. (A) в файлі виводу стандартної ліжком, пептиди, зіставлення екзона відображаються у вигляді єдиного блоку (зліва), в той час як пептиди, зіставлення через кілька екзонів голи екзона, що охоплюють частин як блоки (праворуч). Інтронів показуються тонкі лінії об'єднання. Пого колір-коди унікальність зіставлення або пептиди генів, і стенограми з використанням 3-рівневої системи. (B) крім блочну структуру формату ліжка, ліжко PTM виведення підкреслює позицію стовп-поступальні зміни блоків товщиною. Наявність єдиного PTM типу наголошує на зміні амінокислотних залишків з товщиною блоку, в той час як кілька сайтів же PTM об'єднуються в довгі блоки, що охоплюють від першої до останньої модифікації сайту. Пептид зіставлень далі розділені PTM тип і колір кодека на основі модифікації. Будь ласка, натисніть тут, щоб подивитися велику версію цієї фігури.

Малюнок 9. Відстежувати хаб Перегляд в браузері геномі UCSC колоректального раку протеома і phosphoproteome даних. Трек концентратора включає в себе весь протеома даних, а також phosphoproteome. У той час як червоний колір в протеома і phosphoproteome треків вказують унікальність зіставлення одного запис SFN, треки, що закінчується в _ptm Показати сайти фосфорилювання всередині пептиди. Тут червоний колір вказує тип зміни як фосфорилювання. Було виявлено лише два пептиди з кожним показані один фосфорилювання (блоків товщиною). Будь ласка, натисніть тут, щоб подивитися велику версію цієї фігури.

Малюнок 10. Форма phosphopeptides колоректального раку і пов'язані кількісний в IGV. На малюнку показано підмножина 50 рак клітинних ліній. Він Крім того показує чотири колони блоків в різних відтінків світла червоний. Колір вказує відносне достаток від низького (білий) до високого (червоний). У той час як чотири колони спочатку може привести до вважаю, що є 4 пептиди, стає ясно, з пов'язаний на основі послідовності GTF вихідного файлу що вони насправді два пептиди, кожен охоплюють сплайс-Джанкшн. Будь ласка, натисніть тут, щоб подивитися велику версію цієї фігури.
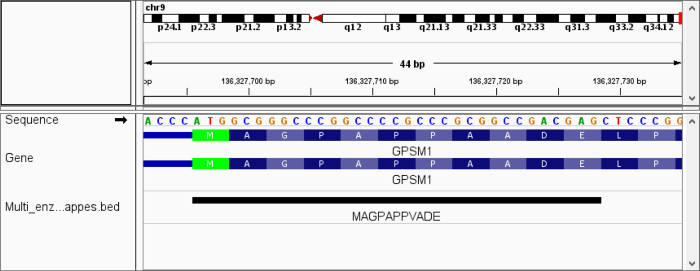
Малюнок 11. Перегляд пептиду з варіантом амінокислоти в IGV. На малюнку показана пептид з однієї амінокислоти варіант зіставляється геном посилання на початку переклад гена GPSM1. Варіант позиціонується на амінокислотний залишок 8 і результати при заміщенні аланина валина (A → V). Послідовності перекладу анотованих стенограми (синій) виділити варіант в порівнянні з пептидного послідовності. Будь ласка, натисніть тут, щоб подивитися велику версію цієї фігури.
